Step-by-Step Guide to Compliance in Medical Device Manufacturing

Following rules is very important in medical device manufacturing. It keeps products safe, high-quality, and ready for global sales. Breaking these rules can cause big problems. These include large fines, expensive recalls, and approval delays. Such issues stop work, waste money, and hurt reputations. Trust is key in healthcare, and breaking rules loses customer trust. It also makes investors lose confidence. To handle medical device manufacturing rules well, you need a clear plan. This helps makers follow rules, avoid problems, and stay successful.
Key Takeaways
Knowing and following medical device rules is very important. It keeps products safe and builds trust with customers.
Create a strong Quality Management System (QMS) to follow the law. A good QMS makes products better and meets rules.
Sort your medical devices by risk level to know approval steps. High-risk devices need more checks and paperwork.
Watching devices after they are sold is very important. It finds problems early and keeps products safe and legal.
Learn about new rules to avoid big mistakes. Regular training and expert help can guide your team with compliance.
Understanding Medical Device Standards and Rules
Knowing medical device rules is key to safety and quality. These rules guide you to meet the needed compliance steps.
FDA Rules (21 CFR Part 820 and QSR)
The FDA has strict rules to keep devices safe. The Quality System Regulation (QSR) in 21 CFR Part 820 helps companies set up a quality system. You must follow these rules to meet FDA standards. This includes proper design, making, and checking after sales.
Over half of medical device companies say new rules cost more. But following these rules lowers risks and builds customer trust. Meeting FDA rules helps avoid fines and sell in the U.S.
ISO 13485: Quality System for Medical Devices
ISO 13485 is a global rule for quality systems in medical devices. It focuses on safety and meeting rules. Following ISO means managing risks, keeping records, and improving processes.
Benefits of ISO standards include:
Success Metric | Description |
|---|---|
Customer Trust | Certification shows quality and rule-following, making customers feel safe. |
Better Processes | Following rules helps reduce waste and improve work. |
Risk Control | Fixing problems early leads to better choices and fewer issues. |
Market Growth | Meeting rules opens new markets, boosting sales and income. |
Global Standards | Following global rules improves reputation and competition. |
Following ISO rules helps you work better and sell in more places.
CE Marking and EU MDR Rules
To sell in Europe, you must follow EU Medical Device Regulation (MDR). CE marking shows your product is safe and works well. To get this, you need documents, clinical checks, and a system to track products after sales.
The medical device market is growing fast with new tech and quicker approvals. Keeping up with EU MDR rules helps you stay competitive in Europe.
Regional Standards in Japan, Canada, and Australia
Knowing regional rules is key for global medical device sales. Each country has its own rules to ensure safety and quality. Let’s look at the main rules in Japan, Canada, and Australia.
Japan: PMDA and MHLW Rules
Japan's medical device rules are managed by PMDA and MHLW. You must follow the PMD Act, which ensures safety, quality, and performance.
Main points to know:
Pre-Market Approval: Devices need approval based on their risk level.
QMS Compliance: Japan has its own QMS rules, similar to ISO 13485 but with extra local needs.
Language Requirements: All papers must be written in Japanese.
Tip: Working with a local expert can make approvals easier and help meet Japan's rules.
Canada: Health Canada and MDSAP
Health Canada controls devices under the Medical Devices Regulations. Devices must be classified, and high-risk ones need an MDL.
Key details include:
MDSAP Certification: Canada uses MDSAP to simplify audits for many markets.
Bilingual Labeling: Labels must be in English and French.
Post-Market Obligations: Report problems and keep records to stay compliant.
Australia: TGA and ARTG Listing
Australia's TGA oversees medical device rules. Devices must be listed in the ARTG before being sold.
Important rules:
Risk-Based Classification: Devices are grouped into four risk levels.
Conformity Assessment: Show compliance with key principles like ISO 13485.
Local Sponsor: A local sponsor must represent your company in Australia.
Note: Following TGA rules can also help you enter other Asia-Pacific markets.
By learning these rules, you can handle regulations better and grow your business.
Step 1: Group Your Medical Device
What is Medical Device Grouping?
Grouping your medical device is the first step to follow rules. Regulators use systems to sort devices by use and patient risks. This step decides how much checking your product needs for approval.
Most places use levels to group devices. For example, the U.S. has Class I, II, and III. Europe and others use Class I, IIa, IIb, and III. Class I devices, like bandages, are low-risk. Class III devices, like pacemakers, are high-risk and need more tests and papers.
Tip: Always check the grouping rules for your market. A low-risk device in one place might be higher-risk elsewhere.
Risk Levels and Rule Steps
The risk level of your device decides the steps to follow. Higher-risk devices need stricter checks, like trials and detailed papers. Knowing these steps helps you plan better.
Here’s a table comparing risk groups in different places:
Place | Risk Groups | Details |
|---|---|---|
EU | I, IIa, IIb, III | Class I is low risk; Class III is high risk. Software depends on use. |
UK | Class I (old EU MDD) | Until 2027, UK uses old EU rules. More software is in Class I. |
US | Class I, II, III | Class I has less checks; Class III needs strict controls. |
Most places use similar risk levels. For example, Mexico, Europe, and Australia use Class I to III. India uses Class A to D. The chart below shows risk levels in different places:
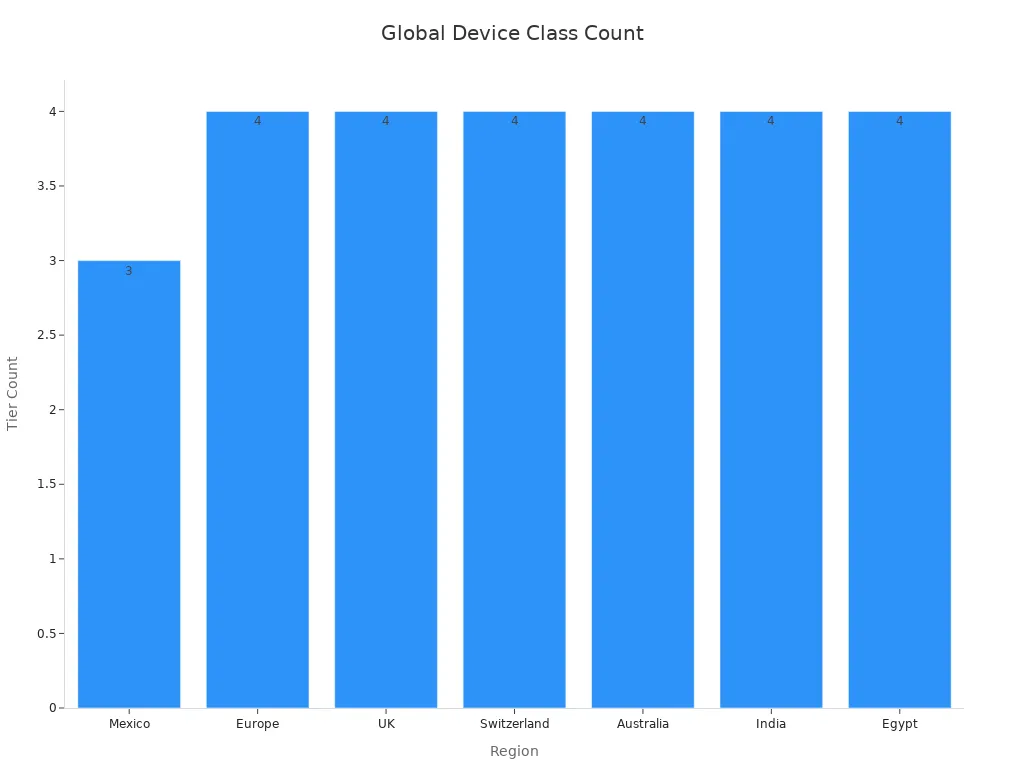
Note: Wrongly grouping your device can cause delays and cost more. Ask experts to get it right.
By learning about device groups and risks, you can follow rules easily and avoid problems.
Step 2: Set Up a Quality Management System (QMS)
A Quality Management System (QMS) is key for following medical device rules. It helps make sure your processes meet the law and create safe products. A good QMS improves both compliance and product quality.
Important Parts of a QMS
To build a strong QMS, focus on its main parts. These parts help your system meet quality and legal standards:
Document Control: Keep clear and updated records to show compliance.
Risk Management: Find and fix risks to keep products safe.
Corrective and Preventive Actions (CAPA): Solve problems and stop them from happening again.
Supplier Quality Management: Check suppliers to ensure they meet your standards.
Internal Audits: Review your processes often to find ways to improve.
You can measure how well these parts work using metrics like customer feedback and supplier performance. Here's a summary:
Metric | What It Shows |
|---|---|
Customer feedback | What customers think about product quality and service. |
Supplier performance | How well suppliers meet your quality needs. |
Product and process monitoring | Checks to find areas that need improvement. |
Non-conformances | Problems where products or processes fail to meet standards. |
Trends | Patterns in data that help improve quality. |
Corrective and preventive actions | Steps taken to fix issues and stop them from happening again. |
Tips for Keeping Records
Good record-keeping is important for meeting rules and getting certified. It proves you follow quality system rules. Use these tips to organize your records:
Arrange documents neatly for easy access during checks.
Know which records are needed and check they meet rules.
Work with experts to make sure records are correct and complete.
Groups like the FDA and EU stress using enough samples for testing and supplier checks. Keeping good records of these tests reduces mistakes and helps meet rules.
Tip: Well-organized records make audits easier and leave a good impression.
Training and Audits
Training and audits are vital for staying compliant. Teach your team what they need to know about rules and quality standards. Focus on these areas:
Employee Training: Train workers for their specific roles and duties.
Audit Preparation: Do regular audits to find and fix problems early.
Continuous Improvement: Use audit results to make processes better.
Using data, like sample sizes, makes testing and problem-solving more reliable. This helps meet rules and keeps improving your system.
Note: Regular training and audits build a strong quality culture and keep your team ready for inspections.
Step 3: Create Technical Documentation
Making technical documents is an important part of compliance. These documents prove your device is safe and meets the rules. A good technical file can speed up approvals and avoid delays.
Main Parts of a Technical File
Your technical file must have key details about your device. These details show it follows the rules. Important parts include:
Device Description: Explain what your device does and how it works.
Design and Manufacturing Info: Add drawings, specs, and how it’s made.
Risk Management Records: Show how you found and fixed risks.
Clinical Data: Provide proof your device is safe and works well.
Post-Market Plan: Share how you’ll check the device after selling it.
Tip: Keep your file neat and clear. This helps regulators review it faster.
Managing Risks and User Safety
Risk management keeps your device safe for people to use. Find dangers, check their effects, and fix them. Use tools like FMEA to study risks step by step.
User safety focuses on how people use your device. Test it to find design problems that might cause mistakes. Fixing these early makes it safer and easier to use.
Note: Regulators like devices with strong risk and safety plans.
Labels and User Instructions (IFU)
Good labels and instructions help people use devices safely. Labels should include:
Device name and type
Maker’s information
Warnings and safety tips
Expiration date, if needed
The Instructions for Use (IFU) should explain how to use, clean, and throw away the device. Use simple words and pictures to make it easy to follow.
Reminder: Bad labels can confuse users and cause harm. Test your labels and IFU with real users to ensure they are clear.
Step 4: Perform Clinical Evaluations
Clinical evaluations show that your medical device is safe and works well. This step involves gathering, studying, and sharing clinical data to meet rules and protect patients.
Collecting Clinical Data
Collecting clinical data is very important for following rules. It proves your device meets ethical and scientific standards. Following Good Clinical Practice (GCP) keeps patients safe and ensures good data. Key points include:
GCP protects participants' rights and safety.
Correct data helps with approvals and meeting rules.
Using statistics gives reliable results for better decisions.
Choose the right sample size for your study. A good sample size avoids wrong results and shows how your device works in real life. Collecting proper data proves your device is safe and builds trust with regulators.
Planning and Running Clinical Studies
A good clinical study plan helps get regulatory approval. Start by setting clear goals and picking the right methods. Agencies like the FDA need strong clinical proof for high-risk devices. For example:
Premarket Approval (PMA) needs a lot of clinical data.
Medium-risk devices may need studies for 510(k) approval.
Following GCP rules ensures your study is credible and meets standards.
Your study plan should explain how you will analyze and report data. This shows your device is safe and works well, making approval easier.
Post-Market Clinical Follow-Up (PMCF)
PMCF checks your device’s safety and performance after it’s sold. It collects real-world data to find risks and improve safety. Benefits of PMCF include:
Key Point | Explanation |
|---|---|
Better Patient Safety | Finding problems early through feedback protects users. |
Improved Device Design | Real-world data helps fix issues and make devices safer. |
Stronger Medical Evidence | PMCF adds real-world data to pre-market studies for a full view. |
Market Trust and Responsibility | PMCF shows you care about safety and builds customer trust. |
Meeting Regulatory Rules | PMCF is often required to keep selling your device and support new claims. |
Doing PMCF ensures your device stays safe and meets rules. It also helps improve your product and keeps it competitive in the market.
Step 5: Get Regulatory Approvals
Getting regulatory approval is very important for medical devices. It shows your device meets rules and is safe to use. Each region has its own rules and steps for approval. Knowing these steps helps you move through them faster.
FDA Approval Paths (510(k), PMA, De Novo)
The FDA has three main ways to approve devices: 510(k), PMA, and De Novo. The path depends on your device's risk and how new it is.
510(k) Path: For devices like ones already sold. Approval takes about 177 days. Only 19% get cleared in under three months.
PMA (Premarket Approval): Needed for high-risk devices. This takes about 243 days, with a 92% success rate.
De Novo Path: For new devices with no similar ones. It takes about 338 days, but times can vary from one month to over 30 months.
Pick the right path based on your device's type and needs. Good planning can save time and improve approval chances.
CE Marking and EU MDR Rules
To sell in Europe, you need CE marking under EU MDR. Follow these steps:
Step | What to Do |
|---|---|
1 | Check your device meets MDR safety and performance rules. |
2 | Confirm you follow EU rules as the legal maker. |
3 | Write an EU Declaration of Conformity (DoC). |
4 | Get certification from a Notified Body for higher-risk devices. |
5 | Receive CE marking for your QMS and technical documents. |
The EU MDR adds new rules like better tracking and safety checks after sales. These changes keep devices safe and working well.
Regional Approval in Japan, Canada, and Australia
Each region has its own approval rules.
Japan: Follow PMD Act rules. Submit papers in Japanese and meet local QMS standards.
Canada: Classify devices. High-risk ones need an MDL. MDSAP makes audits easier for many markets.
Australia: List devices in the ARTG. Use risk-based groups and show compliance with key rules.
Knowing these regional rules helps you get approvals faster and sell in more places.
Tip: Work with local experts to handle tricky rules and avoid delays.
Step 6: Post-Market Surveillance and Reporting Obligations
Post-market surveillance checks if your device stays safe and works well. This step is key to following rules and meeting ongoing needs.
Why Post-Market Surveillance Matters
Post-market surveillance helps find and fix safety problems. It uses real-world data to improve devices and meet rules. Here’s how it helps:
Evidence Type | Description |
|---|---|
Early Problem Detection | Finds safety issues early for faster fixes. |
Better Device Design | Shows areas to improve for safer devices. |
Real-World Insights | Gives data on how devices work outside labs. |
Smarter Decisions | Helps doctors make better choices. |
Maker Responsibility | Proves you care about safety and quality. |
User Trust | Builds trust with patients and doctors through openness. |
Rule Compliance | Keeps your device approved for sale. |
Focusing on post-market surveillance boosts safety, improves devices, and builds trust with users and regulators.
Reporting Problems and Vigilance Systems
Reporting problems is a big part of post-market surveillance. You must track and report unexpected device effects. This helps find risks and keeps users safe. Vigilance systems study patterns in problems to fix issues early.
Aspect | Description |
|---|---|
Problem Reporting | Tracks and reports unexpected effects for safety. |
Pattern Analysis | Finds trends and risks for early improvements. |
A strong vigilance system meets rules and shows your focus on safety and quality.
Improving and Updating Compliance
Improving is key to staying ahead in medical device rules. Measuring performance helps improve your Quality Management System (QMS). Use these steps to stay compliant and improve:
Pick Key Performance Indicators (KPIs) to track quality and find weak spots.
Fix problems with corrective actions and stop future ones with preventive steps.
Follow the Plan-Do-Check-Act (PDCA) cycle:
Plan: Spot areas to improve and make plans.
Do: Test changes on a small scale.
Check: Review results to see if they worked.
Act: Keep good changes or adjust plans and try again.
By improving constantly, you can handle new rules, boost safety, and stay compliant.
Common Challenges in Medical Device Compliance
Understanding Complicated Rules
Following rules for medical devices can be hard. Each place has its own rules. Breaking them can cause delays, fines, or recalls. Teams often don’t fully understand these rules. This can lead to mistakes and approval delays. Not enough workers make things worse by slowing progress. Protecting patient data while following laws like HIPAA is also tough. These problems show why a trained team and clear plan are important.
Handling Costs and Resources
Meeting rules can cost a lot of money. Companies must pay for quality checks, studies, and documents. Breaking rules costs even more, like fines or recalls. For example, not meeting standards can stop production or reject products. Delays in approval hurt sales and growth. To save money, focus on risky areas and train workers. Planning ahead helps avoid mistakes and speeds up approvals.
Keeping Up with Changing Rules
Medical device rules change often to stay safe with new tech. Staying updated is hard but important. Breaking new rules can hurt your reputation and lose customer trust. Bad news about safety can scare investors and harm your business. To stay updated, watch for rule changes and adjust your work. Experts or software can help you follow the latest rules. This keeps your devices safe and approved.
Following steps helps keep medical devices safe and high-quality. Group your device, set up a strong quality system, and make clear technical files. These actions meet rules and avoid fines or delays. They also protect your company’s reputation and build trust with customers and regulators.
Stay ready for new rules by training workers and doing audits often. Experts can guide you through the process. This makes it easier to follow rules and sell safe, dependable products.
FAQ
What is the key step for medical device compliance?
All steps matter, but setting up a Quality Management System (QMS) is key. It ensures your processes follow rules and make safe devices. A good QMS also helps manage risks, improve quality, and get ready for audits.
How can I find my medical device's classification?
You can figure out your device's class by checking its use and risk. Look at the rules for your market, like FDA or EU MDR. Asking an expert can help you avoid mistakes in classification.
Why do we need post-market surveillance?
Post-market surveillance checks if your device stays safe and works well after being sold. It finds real-world problems, improves designs, and keeps you following rules. Regulators often require it to keep selling your product.
Do all medical devices need clinical studies?
Not every device needs clinical studies. Low-risk devices might only need existing data or research reviews. High-risk devices usually need trials to prove safety and how well they work. Always check the rules for your device type.
How can I keep up with changing rules?
Sign up for rule updates, join webinars, and follow groups like the FDA or ISO. Using compliance tools or hiring experts can also help you stay updated and adjust quickly.
See Also
Comprehensive Process Overview for Microcatheter Production
Understanding Insulation Barriers for Medical Device Standards
Detailed Instructions for Nitinol Microtubing Production in Neurovascular Use
Selecting Optimal FEP Tubing for Medical Device Applications

© Copyright 2024 My First Blog - All Rights Reserved.